
今天上午,全球顶级医学期刊《新英格兰医学杂志》在线发表了口服核苷类抗新型冠状病毒药物VV116对比奈玛特韦片/利托那韦片组合药物(PAXLOVID)用于伴有进展为重度(包括死亡高风险因素)的轻至中度新冠病毒感染患者早期治疗的Ⅲ期临床研究成果。VV116是我国科学家自主研发的一款抗新冠病毒口服药,目前处于国际多中心Ⅲ期临床研究阶段。
这项研究由上海交通大学医学院附属瑞金医院牵头开展,是奥密克戎变异株流行期间首个针对中国新冠感染者开展的小分子口服抗病毒药物“头对头”Ⅲ期临床研究。结果显示,研究主要终点达到设计的非劣效终点,相比PAXLOVID,VV116组的临床恢复时间更短,安全性方面的顾虑更少。

《新英格兰医学杂志》论文截图
我国自主研发新药得到国际认可
中国工程院院士、瑞金医院院长宁光表示:“这项发表在《新英格兰医学杂志》上的论文研究结果证实VV116的临床疗效不亚于PAXLOVID。如今中国疫情政策转向,国门即将打开,我们这项研究不仅为全球抗新冠小分子药物的研发和临床应用提供了宝贵的数据和经验,也将为中国抗疫事业做出贡献。”
据介绍,VV116是一种RdRp(核糖核酸聚合酶)抑制剂,可抑制新冠病毒在人体内的复制。它由中国科学院上海药物研究所、中国科学院武汉病毒研究所、中国科学院新疆理化技术研究所、中国科学院中亚药物研发中心/中乌医药科技城(科技部“一带一路”联合实验室)、临港实验室、旺山旺水和君实生物共同研发。君实生物与旺山旺水共同承担该药物在全球层面的临床开发和产业化工作。
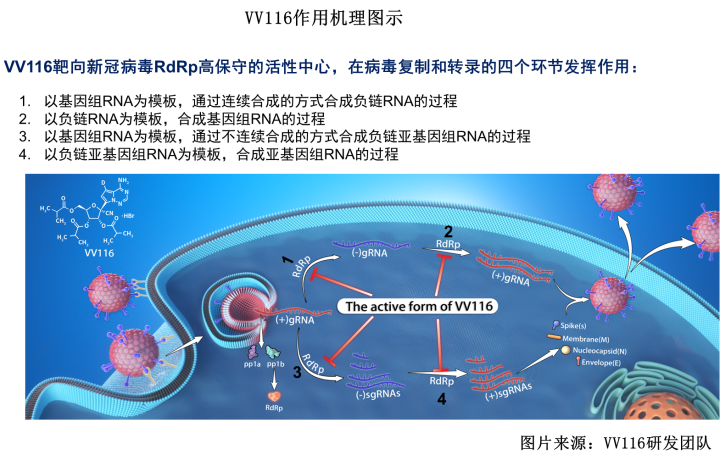
君实生物全球研发总裁邹建军博士说:“这项研究在《新英格兰医学杂志》的发表,证明了国际学术界对中国专家、中国制药企业共同主导的药物开发给予了高度认可。我们正在持续投入VV116在其他人群中的适应症的临床开发工作,希望能够通过这款新疗法为我国乃至全球新冠患者提供效果更好、更安全的治疗选择。”
临床前研究显示,VV116对新冠病毒原始株和已知突变株都表现出显著的抗病毒作用,并在Ⅰ期临床研究中表现出令人满意的安全性、耐受性和药代动力学性质。一项初步的小规模研究证实,与常规治疗相比,首次核酸检测为阳性后5天内接受VV116治疗,患者的核酸转阴时间更短。
与PAXLOVID进行多项指标对照
今天在《新英格兰医学杂志》上发表的是一项多中心、单盲(研究者保持盲态)、随机、对照Ⅲ期临床试验,于今年4月4日至5月2日,在上海的7家新冠肺炎定点医院联合开展,共纳入822例确诊为伴有进展高风险的轻度至中度成人患者,按照1:1的比例被分配至VV116组和PAXLOVID组。最终,共有771例患者接受了VV116或PAXLOVID的治疗。
这些患者的中位年龄为53岁(范围:18—94岁),女性占比50.2%,轻症患者占比92.1%,75.7%的患者全程接种新冠疫苗或接种过加强针,77.3%的患者在症状出现5天内接受了VV116或PAXLOVID治疗。患者中最常见的高风险因素包括:年龄≥60岁(37.7%)、心血管疾病(包括高血压)(35.1%)、肥胖或超重BMI≥25(32.9%)、目前吸烟(12.5%)和糖尿病(10.1%)。
研究的主要终点是从随机至持续临床恢复的时间,风险比两侧95%置信区间下限>0.8定义为非劣效性。次要疗效终点包括截至第28天进展为重度/危重或全因死亡的患者比例,新冠病毒感染相关症状评分和WHO临床进展量表评分变化、至持续症状消失的时间、核酸阴性时间等。安全性终点包括不良事件和严重不良事件情况。
根据最终分析结果(截至今年8月18日),在受试患者中,VV116与PAXLOVID在“至持续临床恢复的时间”达到非劣效,且VV116组比PAXLOVID组的中位恢复时间更短。
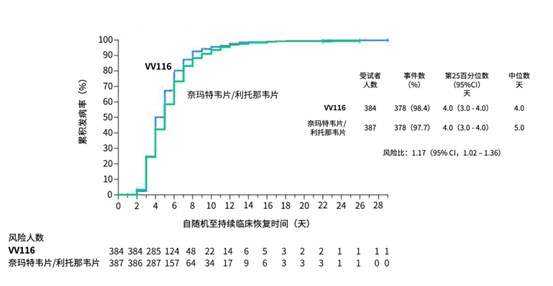
受试人群的至持续临床恢复时间最终分析结果
VV116组和PAXLOVID组在“至持续症状消失的时间”“至首次核酸阴性时间”方面表现类似,中位时间均为7天。在每一个预设时间点(第5、7、10、14、28天),VV116组症状缓解的患者比例均高于PAXLOVID组。两组患者均未发生进展为重度/危重或死亡。
在安全性方面,VV116比PAXLOVID的安全性顾虑更少。VV116组的不良事件发生率低于PAXLOVID组。值得注意的是,PAXLOVID与多种药物存在相互作用,而VV116不会抑制或诱导主要药物代谢酶,也不会抑制主要药物转运蛋白,所以与合并用药发生相互作用的可能性很小。


